Learning Molecular Representation using Graph Neural Network - Training Molecular Graph
Taking a look at how graph neural network operate for molecular representations



Motivation
In the previous post, I have looked into how a molecular graph is constructed and message can be passed around in a MPNN architecture. In this post, I'll take a look at how the graph neural net can be trained. The details of message passing update will vary by implementation; here we choose what was used in this paper.
Again, many code examples were taken from chemprop repository. The code was initially taken from the chemprop repository and I edited them for the sake of simplicity.
Recap
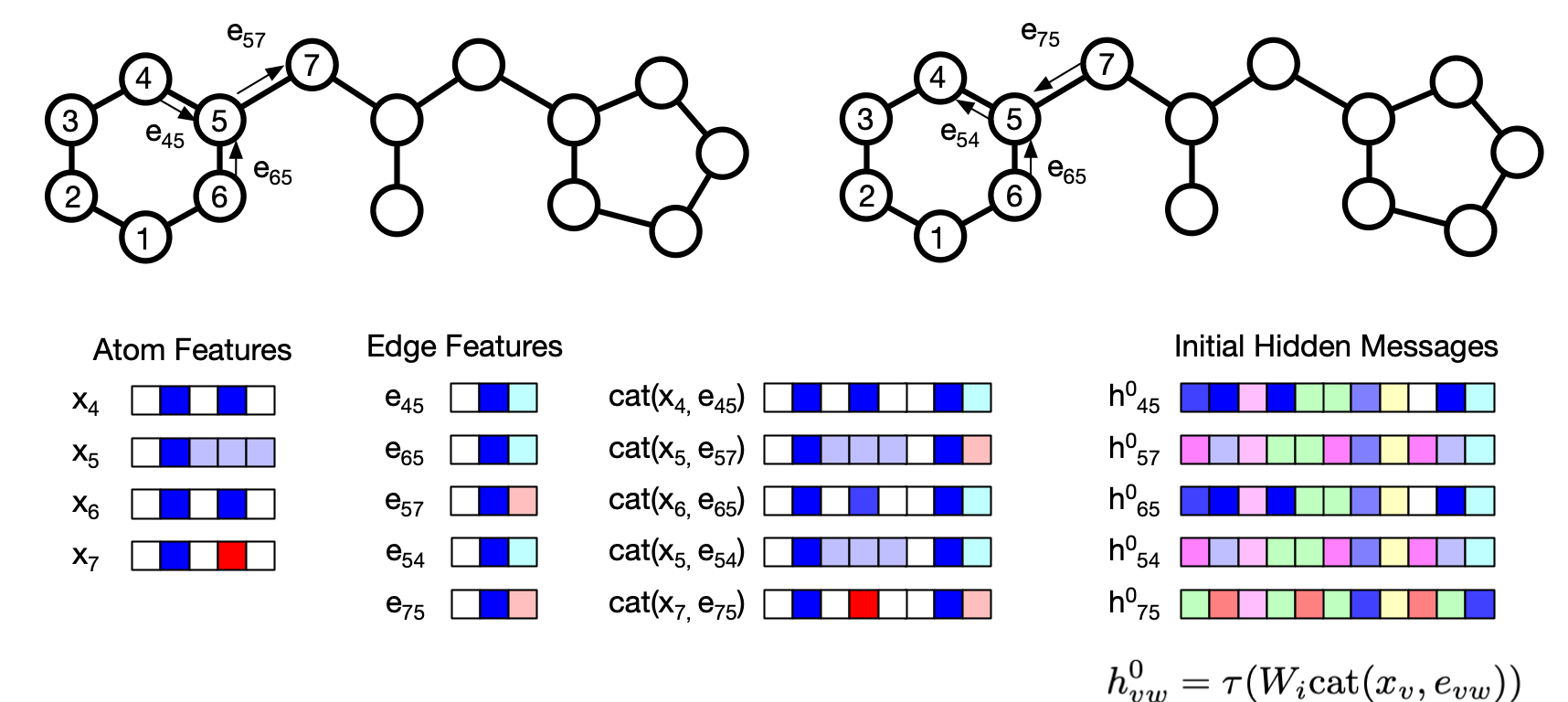
Let's briefly recap how the molecular graph is constructed and messages are passed around. As shown in the above figure, each atom and bonds are labeled as $x$ and $e$. Here we are discussing D-MPNN, which represents the graph with a directional edges, which means, for each bond between two atoms $v$ and $w$, there are two directional bond, $e_{vw}$ and $e_{wv}$.
The initial hidden message is constructed as $h_{vw}^0 = \tau (W_i \mathrm{cat}(x_v, e_{vw}))$ where $W_i$ is a learned matrix and $\tau$ is an activation function.
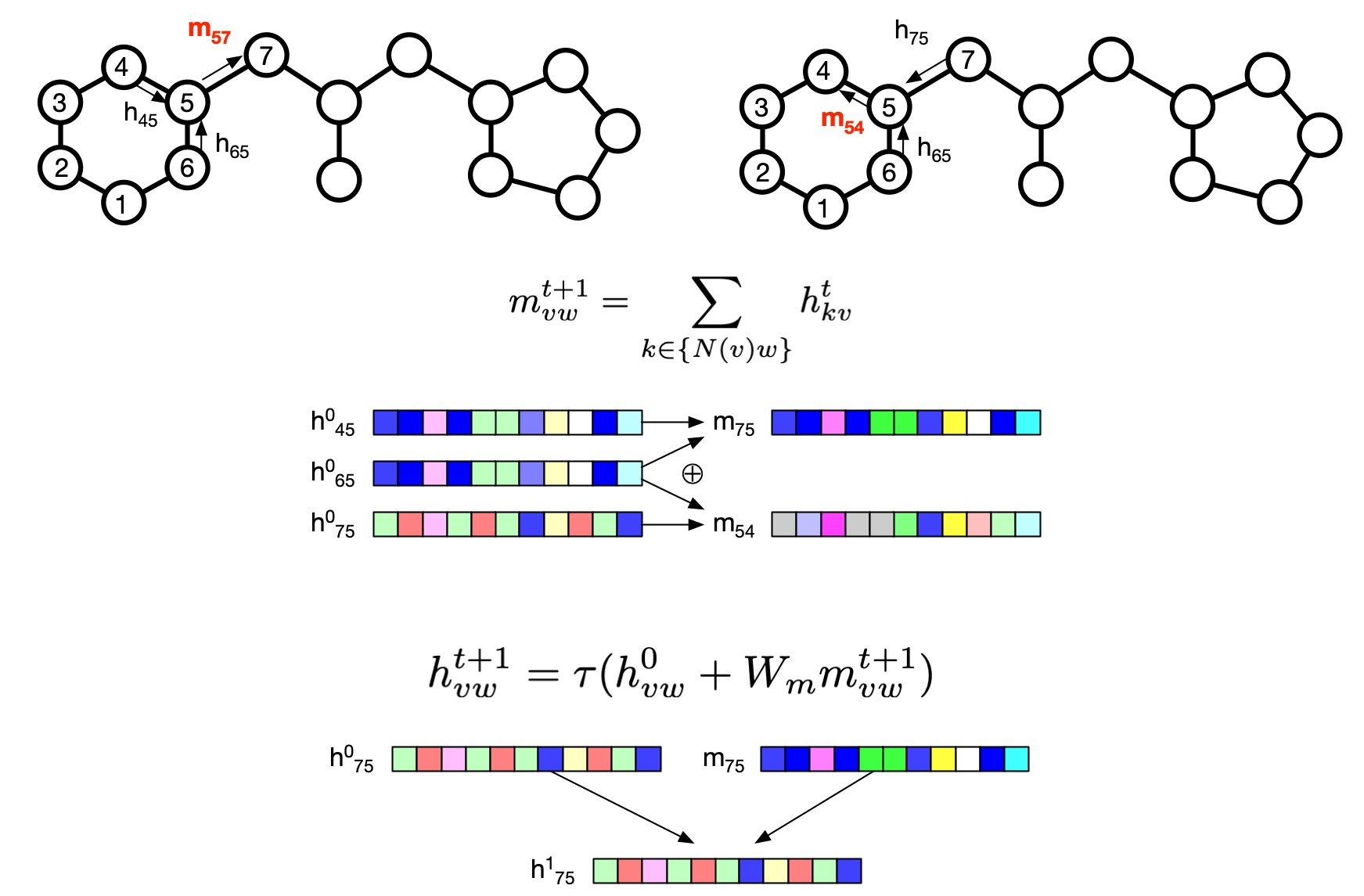
Above figure shows two messages, $m_{57}$ and $m_{54}$, are constructed. First, the message from atom $v$ to $w$ is the sum of all the hidden state for the incoming bonds to $v$ (excluding the one originating from $w$). Then the learned matrix $W_m$ is multiplied to the message and the initial hidden message is added to form the new hidden message for the depth 1. This is repeated several times for the message to be passed around to multiple depth.
After the messages are passed up to the given number of depth, the hidden states are summed to be a final message per each atom (all incoming hidden state) and the hidden state for each atom is computed as follows:
$$m_v = \sum_{k \in N(v)} h_{kv}^t$$
$$h_v = \tau(W_a \mathrm{cat} (x_v, m_v))$$
Finally, the readout phase uses the sum of all $h_v$ to obtain the feature vector of the molecule and property prediction is carried out using a fully-connected feed forward network.
%matplotlib inline
import matplotlib.pyplot as plt
import matplotlib
from io import BytesIO
import pandas as pd
import numpy as np
from IPython.display import SVG
# RDKit
import rdkit
from rdkit.Chem import PandasTools
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import DataStructs
from rdkit.Chem import rdMolDescriptors
from rdkit.Chem import rdRGroupDecomposition
from rdkit.Chem.Draw import IPythonConsole #Needed to show molecules
from rdkit.Chem import Draw
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import rdMolDraw2D
from rdkit.Chem.Draw.MolDrawing import MolDrawing, DrawingOptions #Only needed if modifying defaults
DrawingOptions.bondLineWidth=1.8
IPythonConsole.ipython_useSVG=True
from rdkit import RDLogger
RDLogger.DisableLog('rdApp.warning')
print(rdkit.__version__)
# pytorch
import torch
from torch.utils.data import DataLoader, Dataset, Sampler
from torch import nn
# misc
from typing import Dict, Iterator, List, Optional, Union, OrderedDict, Tuple
from tqdm.notebook import tqdm
from functools import reduce
# we will define a class which holds various parameter for D-MPNN
class TrainArgs:
smiles_column = None
no_cuda = False
gpu = None
num_workers = 8
batch_size = 50
no_cache_mol = False
dataset_type = 'regression'
task_names = []
seed = 0
hidden_size = 300
bias = False
depth = 3
dropout = 0.0
undirected = False
aggregation = 'mean'
aggregation_norm = 100
ffn_num_layers = 2
ffn_hidden_size = 300
init_lr = 1e-4
max_lr = 1e-3
final_lr = 1e-4
num_lrs = 1
warmup_epochs = 2.0
epochs = 30
@property
def device(self) -> torch.device:
"""The :code:`torch.device` on which to load and process data and models."""
if not self.cuda:
return torch.device('cpu')
return torch.device('cuda', self.gpu)
@device.setter
def device(self, device: torch.device) -> None:
self.cuda = device.type == 'cuda'
self.gpu = device.index
@property
def cuda(self) -> bool:
"""Whether to use CUDA (i.e., GPUs) or not."""
return not self.no_cuda and torch.cuda.is_available()
@cuda.setter
def cuda(self, cuda: bool) -> None:
self.no_cuda = not cuda
args = TrainArgs()
args.data_path = 'files/enamine_discovery_diversity_set_10240.csv'
args.target_column = 'ClogP'
args.smiles_column = 'SMILES'
args.dataset_type = 'regression'
args.task_names = [args.target_column]
args.num_tasks = 1
df = pd.read_csv(args.data_path)
df.head()
from random import Random
# Cache of graph featurizations
CACHE_GRAPH = True
SMILES_TO_GRAPH = {}
def cache_graph():
return CACHE_GRAPH
def set_cache_graph(cache_graph):
global CACHE_GRAPH
CACHE_GRAPH = cache_graph
# Cache of RDKit molecules
CACHE_MOL = True
SMILES_TO_MOL: Dict[str, Chem.Mol] = {}
def cache_mol() -> bool:
r"""Returns whether RDKit molecules will be cached."""
return CACHE_MOL
def set_cache_mol(cache_mol: bool) -> None:
r"""Sets whether RDKit molecules will be cached."""
global CACHE_MOL
CACHE_MOL = cache_mol
# Atom feature sizes
MAX_ATOMIC_NUM = 100
ATOM_FEATURES = {
'atomic_num': list(range(MAX_ATOMIC_NUM)),
'degree': [0, 1, 2, 3, 4, 5],
'formal_charge': [-1, -2, 1, 2, 0],
'chiral_tag': [0, 1, 2, 3],
'num_Hs': [0, 1, 2, 3, 4],
'hybridization': [
Chem.rdchem.HybridizationType.SP,
Chem.rdchem.HybridizationType.SP2,
Chem.rdchem.HybridizationType.SP3,
Chem.rdchem.HybridizationType.SP3D,
Chem.rdchem.HybridizationType.SP3D2
],
}
# Distance feature sizes
PATH_DISTANCE_BINS = list(range(10))
THREE_D_DISTANCE_MAX = 20
THREE_D_DISTANCE_STEP = 1
THREE_D_DISTANCE_BINS = list(range(0, THREE_D_DISTANCE_MAX + 1, THREE_D_DISTANCE_STEP))
# len(choices) + 1 to include room for uncommon values; + 2 at end for IsAromatic and mass
ATOM_FDIM = sum(len(choices) + 1 for choices in ATOM_FEATURES.values()) + 2
EXTRA_ATOM_FDIM = 0
BOND_FDIM = 14
def get_atom_fdim():
"""Gets the dimensionality of the atom feature vector."""
return ATOM_FDIM + EXTRA_ATOM_FDIM
def get_bond_fdim(atom_messages=False):
"""Gets the dimensionality of the bond feature vector.
"""
return BOND_FDIM + (not atom_messages) * get_atom_fdim()
def onek_encoding_unk(value: int, choices: List[int]):
encoding = [0] * (len(choices) + 1)
index = choices.index(value) if value in choices else -1
encoding[index] = 1
return encoding
def atom_features(atom: Chem.rdchem.Atom, functional_groups: List[int] = None):
"""Builds a feature vector for an atom.
"""
features = onek_encoding_unk(atom.GetAtomicNum() - 1, ATOM_FEATURES['atomic_num']) + \
onek_encoding_unk(atom.GetTotalDegree(), ATOM_FEATURES['degree']) + \
onek_encoding_unk(atom.GetFormalCharge(), ATOM_FEATURES['formal_charge']) + \
onek_encoding_unk(int(atom.GetChiralTag()), ATOM_FEATURES['chiral_tag']) + \
onek_encoding_unk(int(atom.GetTotalNumHs()), ATOM_FEATURES['num_Hs']) + \
onek_encoding_unk(int(atom.GetHybridization()), ATOM_FEATURES['hybridization']) + \
[1 if atom.GetIsAromatic() else 0] + \
[atom.GetMass() * 0.01] # scaled to about the same range as other features
if functional_groups is not None:
features += functional_groups
return features
def bond_features(bond: Chem.rdchem.Bond):
"""Builds a feature vector for a bond.
"""
if bond is None:
fbond = [1] + [0] * (BOND_FDIM - 1)
else:
bt = bond.GetBondType()
fbond = [
0, # bond is not None
bt == Chem.rdchem.BondType.SINGLE,
bt == Chem.rdchem.BondType.DOUBLE,
bt == Chem.rdchem.BondType.TRIPLE,
bt == Chem.rdchem.BondType.AROMATIC,
(bond.GetIsConjugated() if bt is not None else 0),
(bond.IsInRing() if bt is not None else 0)
]
fbond += onek_encoding_unk(int(bond.GetStereo()), list(range(6)))
return fbond
class MoleculeDatapoint:
def __init__(self,
smiles: str,
targets: List[Optional[float]] = None,
row: OrderedDict = None):
self.smiles = smiles
self.targets = targets
self.features = []
self.row = row
@property
def mol(self) -> Chem.Mol:
"""Gets the corresponding list of RDKit molecules for the corresponding SMILES."""
mol = SMILES_TO_MOL.get(self.smiles, Chem.MolFromSmiles(self.smiles))
if cache_mol():
SMILES_TO_MOL[self.smiles] = mol
return mol
def set_features(self, features: np.ndarray) -> None:
"""Sets the features of the molecule.
"""
self.features = features
def extend_features(self, features: np.ndarray) -> None:
"""Extends the features of the molecule.
"""
self.features = np.append(self.features, features) if self.features is not None else features
def num_tasks(self) -> int:
"""Returns the number of prediction tasks.
"""
return len(self.targets)
def set_targets(self, targets: List[Optional[float]]):
"""Sets the targets of a molecule.
"""
self.targets = targets
def reset_features_and_targets(self) -> None:
"""Resets the features and targets to their raw values."""
self.features, self.targets = self.raw_features, self.raw_targets
class MoleculeDataset(Dataset):
def __init__(self, data: List[MoleculeDatapoint]):
self._data = data
self._scaler = None
self._batch_graph = None
self._random = Random()
def smiles(self) -> List[str]:
return [d.smiles for d in self._data]
def mols(self) -> List[Chem.Mol]:
return [d.mol for d in self._data]
def targets(self) -> List[List[Optional[float]]]:
return [d.targets for d in self._data]
def num_tasks(self) -> int:
return self._data[0].num_tasks() if len(self._data) > 0 else None
def set_targets(self, targets: List[List[Optional[float]]]) -> None:
assert len(self._data) == len(targets)
for i in range(len(self._data)):
self._data[i].set_targets(targets[i])
def reset_features_and_targets(self) -> None:
for d in self._data:
d.reset_features_and_targets()
def __len__(self) -> int:
return len(self._data)
def __getitem__(self, item) -> Union[MoleculeDatapoint, List[MoleculeDatapoint]]:
return self._data[item]
def batch_graph(self):
if self._batch_graph is None:
self._batch_graph = []
mol_graphs = []
for d in self._data:
mol_graphs_list = []
if d.smiles in SMILES_TO_GRAPH:
mol_graph = SMILES_TO_GRAPH[d.smiles]
else:
mol_graph = MolGraph(d.mol)
if cache_graph():
SMILES_TO_GRAPH[d.smiles] = mol_graph
mol_graphs.append([mol_graph])
self._batch_graph = [BatchMolGraph([g[i] for g in mol_graphs]) for i in range(len(mol_graphs[0]))]
return self._batch_graph
def features(self) -> List[np.ndarray]:
"""
Returns the features associated with each molecule (if they exist).
:return: A list of 1D numpy arrays containing the features for each molecule or None if there are no features.
"""
if len(self._data) == 0 or self._data[0].features is None:
return None
return [d.features for d in self._data]
def index_select_ND(source: torch.Tensor, index: torch.Tensor) -> torch.Tensor:
"""Selects the message features from source corresponding to the atom or bond indices in index.
"""
index_size = index.size() # (num_atoms/num_bonds, max_num_bonds)
suffix_dim = source.size()[1:] # (hidden_size,)
final_size = index_size + suffix_dim # (num_atoms/num_bonds, max_num_bonds, hidden_size)
target = source.index_select(dim=0, index=index.view(-1)) # (num_atoms/num_bonds * max_num_bonds, hidden_size)
target = target.view(final_size) # (num_atoms/num_bonds, max_num_bonds, hidden_size)
return target
class MolGraph:
def __init__(self, mol, atom_descriptors=None):
# Convert SMILES to RDKit molecule if necessary
if type(mol) == str:
mol = Chem.MolFromSmiles(mol)
self.n_atoms = 0 # number of atoms
self.n_bonds = 0 # number of bonds
self.f_atoms = [] # mapping from atom index to atom features
self.f_bonds = [] # mapping from bond index to concat(in_atom, bond) features
self.a2b = [] # mapping from atom index to incoming bond indices
self.b2a = [] # mapping from bond index to the index of the atom the bond is coming from
self.b2revb = [] # mapping from bond index to the index of the reverse bond
# Get atom features
self.f_atoms = [atom_features(atom) for atom in mol.GetAtoms()]
if atom_descriptors is not None:
self.f_atoms = [f_atoms + descs.tolist() for f_atoms, descs in zip(self.f_atoms, atom_descriptors)]
self.n_atoms = len(self.f_atoms)
# Initialize atom to bond mapping for each atom
for _ in range(self.n_atoms):
self.a2b.append([])
# Get bond features
for a1 in range(self.n_atoms):
for a2 in range(a1 + 1, self.n_atoms):
bond = mol.GetBondBetweenAtoms(a1, a2)
if bond is None:
continue
f_bond = bond_features(bond)
self.f_bonds.append(self.f_atoms[a1] + f_bond)
self.f_bonds.append(self.f_atoms[a2] + f_bond)
# Update index mappings
b1 = self.n_bonds
b2 = b1 + 1
self.a2b[a2].append(b1) # b1 = a1 --> a2
self.b2a.append(a1)
self.a2b[a1].append(b2) # b2 = a2 --> a1
self.b2a.append(a2)
self.b2revb.append(b2)
self.b2revb.append(b1)
self.n_bonds += 2
class BatchMolGraph:
"""A `BatchMolGraph` represents the graph structure and featurization of a batch of molecules.
"""
def __init__(self, mol_graphs: List[MolGraph]):
self.atom_fdim = get_atom_fdim()
self.bond_fdim = get_bond_fdim()
# Start n_atoms and n_bonds at 1 b/c zero padding
self.n_atoms = 1 # number of atoms (start at 1 b/c need index 0 as padding)
self.n_bonds = 1 # number of bonds (start at 1 b/c need index 0 as padding)
self.a_scope = [] # list of tuples indicating (start_atom_index, num_atoms) for each molecule
self.b_scope = [] # list of tuples indicating (start_bond_index, num_bonds) for each molecule
# All start with zero padding so that indexing with zero padding returns zeros
f_atoms = [[0] * self.atom_fdim] # atom features
f_bonds = [[0] * self.bond_fdim] # combined atom/bond features
a2b = [[]] # mapping from atom index to incoming bond indices
b2a = [0] # mapping from bond index to the index of the atom the bond is coming from
b2revb = [0] # mapping from bond index to the index of the reverse bond
for mol_graph in mol_graphs:
f_atoms.extend(mol_graph.f_atoms)
f_bonds.extend(mol_graph.f_bonds)
for a in range(mol_graph.n_atoms):
a2b.append([b + self.n_bonds for b in mol_graph.a2b[a]])
for b in range(mol_graph.n_bonds):
b2a.append(self.n_atoms + mol_graph.b2a[b])
b2revb.append(self.n_bonds + mol_graph.b2revb[b])
self.a_scope.append((self.n_atoms, mol_graph.n_atoms))
self.b_scope.append((self.n_bonds, mol_graph.n_bonds))
self.n_atoms += mol_graph.n_atoms
self.n_bonds += mol_graph.n_bonds
self.max_num_bonds = max(1, max(len(in_bonds) for in_bonds in a2b)) # max with 1 to fix a crash in rare case of all single-heavy-atom mols
self.f_atoms = torch.FloatTensor(f_atoms)
self.f_bonds = torch.FloatTensor(f_bonds)
self.a2b = torch.LongTensor([a2b[a] + [0] * (self.max_num_bonds - len(a2b[a])) for a in range(self.n_atoms)])
self.b2a = torch.LongTensor(b2a)
self.b2revb = torch.LongTensor(b2revb)
self.b2b = None # try to avoid computing b2b b/c O(n_atoms^3)
self.a2a = None # only needed if using atom messages
def get_components(self, atom_messages: bool = False) -> Tuple[torch.FloatTensor, torch.FloatTensor,
torch.LongTensor, torch.LongTensor, torch.LongTensor,
List[Tuple[int, int]], List[Tuple[int, int]]]:
return self.f_atoms, self.f_bonds, self.a2b, self.b2a, self.b2revb, self.a_scope, self.b_scope
def get_b2b(self) -> torch.LongTensor:
"""Computes (if necessary) and returns a mapping from each bond index to all the incoming bond indices.
"""
if self.b2b is None:
b2b = self.a2b[self.b2a] # num_bonds x max_num_bonds
# b2b includes reverse edge for each bond so need to mask out
revmask = (b2b != self.b2revb.unsqueeze(1).repeat(1, b2b.size(1))).long() # num_bonds x max_num_bonds
self.b2b = b2b * revmask
return self.b2b
def get_a2a(self) -> torch.LongTensor:
"""Computes (if necessary) and returns a mapping from each atom index to all neighboring atom indices.
"""
if self.a2a is None:
# b = a1 --> a2
# a2b maps a2 to all incoming bonds b
# b2a maps each bond b to the atom it comes from a1
# thus b2a[a2b] maps atom a2 to neighboring atoms a1
self.a2a = self.b2a[self.a2b] # num_atoms x max_num_bonds
return self.a2a
# prepare data set
data = MoleculeDataset([
MoleculeDatapoint(
smiles=row[args.smiles_column],
targets=[row[args.target_column]]
) for i, row in df.iterrows()
])
# split data into train, validation and test set
random = Random()
sizes = [0.8, 0.1, 0.1]
indices = list(range(len(data)))
random.shuffle(indices)
train_size = int(sizes[0] * len(data))
train_val_size = int((sizes[0] + sizes[1]) * len(data))
train = [data[i] for i in indices[:train_size]]
val = [data[i] for i in indices[train_size:train_val_size]]
test = [data[i] for i in indices[train_val_size:]]
train_data = MoleculeDataset(train)
val_data = MoleculeDataset(val)
test_data = MoleculeDataset(test)
# Atom feature sizes
MAX_ATOMIC_NUM = 100
ATOM_FEATURES = {
'atomic_num': list(range(MAX_ATOMIC_NUM)),
'degree': [0, 1, 2, 3, 4, 5],
'formal_charge': [-1, -2, 1, 2, 0],
'chiral_tag': [0, 1, 2, 3],
'num_Hs': [0, 1, 2, 3, 4],
'hybridization': [
Chem.rdchem.HybridizationType.SP,
Chem.rdchem.HybridizationType.SP2,
Chem.rdchem.HybridizationType.SP3,
Chem.rdchem.HybridizationType.SP3D,
Chem.rdchem.HybridizationType.SP3D2
],
}
# Distance feature sizes
PATH_DISTANCE_BINS = list(range(10))
THREE_D_DISTANCE_MAX = 20
THREE_D_DISTANCE_STEP = 1
THREE_D_DISTANCE_BINS = list(range(0, THREE_D_DISTANCE_MAX + 1, THREE_D_DISTANCE_STEP))
# len(choices) + 1 to include room for uncommon values; + 2 at end for IsAromatic and mass
ATOM_FDIM = sum(len(choices) + 1 for choices in ATOM_FEATURES.values()) + 2
EXTRA_ATOM_FDIM = 0
BOND_FDIM = 14
def get_atom_fdim() -> int:
"""Gets the dimensionality of the atom feature vector."""
return ATOM_FDIM + EXTRA_ATOM_FDIM
def get_bond_fdim() -> int:
"""Gets the dimensionality of the bond feature vector.
"""
return BOND_FDIM + get_atom_fdim()
def onek_encoding_unk(value: int, choices: List[int]) -> List[int]:
encoding = [0] * (len(choices) + 1)
index = choices.index(value) if value in choices else -1
encoding[index] = 1
return encoding
def atom_features(atom: Chem.rdchem.Atom, functional_groups: List[int] = None) -> List[Union[bool, int, float]]:
"""Builds a feature vector for an atom.
"""
features = onek_encoding_unk(atom.GetAtomicNum() - 1, ATOM_FEATURES['atomic_num']) + \
onek_encoding_unk(atom.GetTotalDegree(), ATOM_FEATURES['degree']) + \
onek_encoding_unk(atom.GetFormalCharge(), ATOM_FEATURES['formal_charge']) + \
onek_encoding_unk(int(atom.GetChiralTag()), ATOM_FEATURES['chiral_tag']) + \
onek_encoding_unk(int(atom.GetTotalNumHs()), ATOM_FEATURES['num_Hs']) + \
onek_encoding_unk(int(atom.GetHybridization()), ATOM_FEATURES['hybridization']) + \
[1 if atom.GetIsAromatic() else 0] + \
[atom.GetMass() * 0.01] # scaled to about the same range as other features
if functional_groups is not None:
features += functional_groups
return features
def initialize_weights(model: nn.Module) -> None:
"""Initializes the weights of a model in place.
"""
for param in model.parameters():
if param.dim() == 1:
nn.init.constant_(param, 0)
else:
nn.init.xavier_normal_(param)
class MPNEncoder(nn.Module):
def __init__(self, args, atom_fdim, bond_fdim):
super(MPNEncoder, self).__init__()
self.atom_fdim = atom_fdim
self.bond_fdim = bond_fdim
self.hidden_size = args.hidden_size
self.bias = args.bias
self.depth = args.depth
self.dropout = args.dropout
self.layers_per_message = 1
self.undirected = False
self.atom_messages = False
self.device = args.device
self.aggregation = args.aggregation
self.aggregation_norm = args.aggregation_norm
self.dropout_layer = nn.Dropout(p=self.dropout)
self.act_func = nn.ReLU()
self.cached_zero_vector = nn.Parameter(torch.zeros(self.hidden_size), requires_grad=False)
# Input
input_dim = self.bond_fdim
self.W_i = nn.Linear(input_dim, self.hidden_size, bias=self.bias)
w_h_input_size = self.hidden_size
# Shared weight matrix across depths (default)
self.W_h = nn.Linear(w_h_input_size, self.hidden_size, bias=self.bias)
self.W_o = nn.Linear(self.atom_fdim + self.hidden_size, self.hidden_size)
def forward(self, mol_graph):
"""Encodes a batch of molecular graphs.
"""
f_atoms, f_bonds, a2b, b2a, b2revb, a_scope, b_scope = mol_graph.get_components()
f_atoms, f_bonds, a2b, b2a, b2revb = f_atoms.to(self.device), f_bonds.to(self.device), a2b.to(self.device), b2a.to(self.device), b2revb.to(self.device)
input = self.W_i(f_bonds) # num_bonds x hidden_size
message = self.act_func(input) # num_bonds x hidden_size
# Message passing
for depth in range(self.depth - 1):
# m(a1 -> a2) = [sum_{a0 \in nei(a1)} m(a0 -> a1)] - m(a2 -> a1)
# message a_message = sum(nei_a_message) rev_message
nei_a_message = index_select_ND(message, a2b) # num_atoms x max_num_bonds x hidden
a_message = nei_a_message.sum(dim=1) # num_atoms x hidden
rev_message = message[b2revb] # num_bonds x hidden
message = a_message[b2a] - rev_message # num_bonds x hidden
message = self.W_h(message)
message = self.act_func(input + message) # num_bonds x hidden_size
message = self.dropout_layer(message) # num_bonds x hidden
a2x = a2b
nei_a_message = index_select_ND(message, a2x) # num_atoms x max_num_bonds x hidden
a_message = nei_a_message.sum(dim=1) # num_atoms x hidden
a_input = torch.cat([f_atoms, a_message], dim=1) # num_atoms x (atom_fdim + hidden)
atom_hiddens = self.act_func(self.W_o(a_input)) # num_atoms x hidden
atom_hiddens = self.dropout_layer(atom_hiddens) # num_atoms x hidden
# Readout
mol_vecs = []
for i, (a_start, a_size) in enumerate(a_scope):
if a_size == 0:
mol_vecs.append(self.cached_zero_vector)
else:
cur_hiddens = atom_hiddens.narrow(0, a_start, a_size)
mol_vec = cur_hiddens # (num_atoms, hidden_size)
if self.aggregation == 'mean':
mol_vec = mol_vec.sum(dim=0) / a_size
elif self.aggregation == 'sum':
mol_vec = mol_vec.sum(dim=0)
elif self.aggregation == 'norm':
mol_vec = mol_vec.sum(dim=0) / self.aggregation_norm
mol_vecs.append(mol_vec)
mol_vecs = torch.stack(mol_vecs, dim=0) # (num_molecules, hidden_size)
return mol_vecs # num_molecules x hidden
class MPN(nn.Module):
def __init__(self, args, atom_fdim=None, bond_fdim=None):
super(MPN, self).__init__()
self.atom_fdim = atom_fdim or get_atom_fdim()
self.bond_fdim = bond_fdim or get_bond_fdim()
self.device = args.device
self.encoder = MPNEncoder(args, self.atom_fdim, self.bond_fdim)
def forward(self, batch):
"""Encodes a batch of molecules.
"""
if type(batch[0]) != BatchMolGraph:
batch = [mol2graph(b) for b in batch]
encodings = [self.encoder(batch[0])]
output = reduce(lambda x, y: torch.cat((x, y), dim=1), encodings)
return output
class MoleculeModel(nn.Module):
def __init__(self, args, featurizer=False):
super(MoleculeModel, self).__init__()
self.classification = args.dataset_type == 'classification'
self.featurizer = featurizer
self.output_size = args.num_tasks
if self.classification:
self.sigmoid = nn.Sigmoid()
self.create_encoder(args)
self.create_ffn(args)
initialize_weights(self)
def create_encoder(self, args):
self.encoder = MPN(args)
def create_ffn(self, args):
first_linear_dim = args.hidden_size
dropout = nn.Dropout(args.dropout)
activation = nn.ReLU()
# Create FFN layers
if args.ffn_num_layers == 1:
ffn = [
dropout,
nn.Linear(first_linear_dim, self.output_size)
]
else:
ffn = [
dropout,
nn.Linear(first_linear_dim, args.ffn_hidden_size)
]
for _ in range(args.ffn_num_layers - 2):
ffn.extend([
activation,
dropout,
nn.Linear(args.ffn_hidden_size, args.ffn_hidden_size),
])
ffn.extend([
activation,
dropout,
nn.Linear(args.ffn_hidden_size, self.output_size),
])
# Create FFN model
self.ffn = nn.Sequential(*ffn)
def featurize(self, batch, features_batch=None, atom_descriptors_batch=None):
"""Computes feature vectors of the input by running the model except for the last layer.
"""
return self.ffn[:-1](self.encoder(batch, features_batch, atom_descriptors_batch))
def forward(self, batch):
output = self.ffn(self.encoder(batch))
# Don't apply sigmoid during training b/c using BCEWithLogitsLoss
if self.classification and not self.training:
output = self.sigmoid(output)
return output
model = MoleculeModel(args)
model = model.to(args.device)
As it is shown below, the model is comprised of an encoder and FFN. The encoder has three learned matrices and the FFN has 2 fully-connected layers.
model
from torch.optim.lr_scheduler import _LRScheduler
from torch.optim import Adam, Optimizer
class NoamLR(_LRScheduler):
"""
Noam learning rate scheduler with piecewise linear increase and exponential decay.
The learning rate increases linearly from init_lr to max_lr over the course of
the first warmup_steps (where :code:`warmup_steps = warmup_epochs * steps_per_epoch`).
Then the learning rate decreases exponentially from :code:`max_lr` to :code:`final_lr` over the
course of the remaining :code:`total_steps - warmup_steps` (where :code:`total_steps =
total_epochs * steps_per_epoch`). This is roughly based on the learning rate
schedule from `Attention is All You Need <https://arxiv.org/abs/1706.03762>`_, section 5.3.
"""
def __init__(self,
optimizer: Optimizer,
warmup_epochs: List[Union[float, int]],
total_epochs: List[int],
steps_per_epoch: int,
init_lr: List[float],
max_lr: List[float],
final_lr: List[float]):
assert len(optimizer.param_groups) == len(warmup_epochs) == len(total_epochs) == len(init_lr) == \
len(max_lr) == len(final_lr)
self.num_lrs = len(optimizer.param_groups)
self.optimizer = optimizer
self.warmup_epochs = np.array(warmup_epochs)
self.total_epochs = np.array(total_epochs)
self.steps_per_epoch = steps_per_epoch
self.init_lr = np.array(init_lr)
self.max_lr = np.array(max_lr)
self.final_lr = np.array(final_lr)
self.current_step = 0
self.lr = init_lr
self.warmup_steps = (self.warmup_epochs * self.steps_per_epoch).astype(int)
self.total_steps = self.total_epochs * self.steps_per_epoch
self.linear_increment = (self.max_lr - self.init_lr) / self.warmup_steps
self.exponential_gamma = (self.final_lr / self.max_lr) ** (1 / (self.total_steps - self.warmup_steps))
super(NoamLR, self).__init__(optimizer)
def get_lr(self) -> List[float]:
return list(self.lr)
def step(self, current_step: int = None):
if current_step is not None:
self.current_step = current_step
else:
self.current_step += 1
for i in range(self.num_lrs):
if self.current_step <= self.warmup_steps[i]:
self.lr[i] = self.init_lr[i] + self.current_step * self.linear_increment[i]
elif self.current_step <= self.total_steps[i]:
self.lr[i] = self.max_lr[i] * (self.exponential_gamma[i] ** (self.current_step - self.warmup_steps[i]))
else: # theoretically this case should never be reached since training should stop at total_steps
self.lr[i] = self.final_lr[i]
self.optimizer.param_groups[i]['lr'] = self.lr[i]
import threading
def construct_molecule_batch(data):
data = MoleculeDataset(data)
data.batch_graph() # Forces computation and caching of the BatchMolGraph for the molecules
return data
class MoleculeSampler(Sampler):
def __init__(self, dataset, shuffle=False, seed=0):
super(Sampler, self).__init__()
self.dataset = dataset
self.shuffle = shuffle
self._random = Random(seed)
self.positive_indices = self.negative_indices = None
self.length = len(self.dataset)
def __iter__(self):
indices = list(range(len(self.dataset)))
if self.shuffle:
self._random.shuffle(indices)
return iter(indices)
def __len__(self):
return self.length
class MoleculeDataLoader(DataLoader):
def __init__(self,
dataset: MoleculeDataset,
batch_size: int = 50,
num_workers: int = 8,
shuffle: bool = False,
seed: int = 0):
self._dataset = dataset
self._batch_size = batch_size
self._num_workers = num_workers
self._shuffle = shuffle
self._seed = seed
self._context = None
self._class_balance = False
self._timeout = 0
is_main_thread = threading.current_thread() is threading.main_thread()
if not is_main_thread and self._num_workers > 0:
self._context = 'forkserver' # In order to prevent a hanging
self._timeout = 3600 # Just for sure that the DataLoader won't hang
self._sampler = MoleculeSampler(
dataset=self._dataset,
shuffle=self._shuffle,
seed=self._seed
)
super(MoleculeDataLoader, self).__init__(
dataset=self._dataset,
batch_size=self._batch_size,
sampler=self._sampler,
num_workers=self._num_workers,
collate_fn=construct_molecule_batch,
multiprocessing_context=self._context,
timeout=self._timeout
)
@property
def targets(self) -> List[List[Optional[float]]]:
if self._class_balance or self._shuffle:
raise ValueError('Cannot safely extract targets when class balance or shuffle are enabled.')
return [self._dataset[index].targets for index in self._sampler]
@property
def iter_size(self) -> int:
return len(self._sampler)
def __iter__(self) -> Iterator[MoleculeDataset]:
return super(MoleculeDataLoader, self).__iter__()
# Create data loaders
train_data_loader = MoleculeDataLoader(
dataset=train_data,
batch_size=args.batch_size,
num_workers=8,
shuffle=True,
seed=args.seed
)
val_data_loader = MoleculeDataLoader(
dataset=val_data,
batch_size=args.batch_size,
num_workers=8
)
test_data_loader = MoleculeDataLoader(
dataset=test_data,
batch_size=args.batch_size,
num_workers=8
)
# optimizer
params = [{'params': model.parameters(), 'lr': args.init_lr, 'weight_decay': 0}]
optimizer = Adam(params)
# scheduler
scheduler = NoamLR(
optimizer=optimizer,
warmup_epochs=[args.warmup_epochs],
total_epochs=[args.epochs] * args.num_lrs,
steps_per_epoch=len(train_data) // args.batch_size,
init_lr=[args.init_lr],
max_lr=[args.max_lr],
final_lr=[args.final_lr]
)
# loss function
loss_func = nn.MSELoss(reduction='none')
# train loop
model.train()
loss_sum = iter_count = 0
n_iter = 0
for batch in tqdm(train_data_loader, total=len(train_data_loader), leave=False):
mol_batch, target_batch = batch.batch_graph(), batch.targets()
mask = torch.Tensor([[x is not None for x in tb] for tb in target_batch])
targets = torch.Tensor([[0 if x is None else x for x in tb] for tb in target_batch])
# Run model
model.zero_grad()
preds = model(mol_batch)
# Move tensors to correct device
mask = mask.to(preds.device)
targets = targets.to(preds.device)
class_weights = torch.ones(targets.shape, device=preds.device)
loss = loss_func(preds, targets) * class_weights * mask
loss = loss.sum() / mask.sum()
loss_sum += loss.item()
iter_count += 1
loss.backward()
optimizer.step()
if isinstance(scheduler, NoamLR):
scheduler.step()
n_iter += len(batch)
Training for the first epoch is finished. Let's take a look at how well the model predict the ClogP.
model.eval()
initial_preds = []
for batch in tqdm(val_data_loader, disable=False, leave=False):
# Prepare batch
batch: MoleculeDataset
mol_batch = batch.batch_graph()
# Make predictions
with torch.no_grad():
batch_preds = model(mol_batch)
batch_preds = batch_preds.data.cpu().numpy()
# Collect vectors
batch_preds = batch_preds.tolist()
initial_preds.extend(batch_preds)
from sklearn.metrics import mean_squared_error
# valid_preds and valid_targets have shape (num_tasks, data_size)
targets = val_data_loader.targets
metric_func = mean_squared_error
valid_preds = [[] for _ in range(args.num_tasks)]
valid_targets = [[] for _ in range(args.num_tasks)]
for i in range(args.num_tasks):
for j in range(len(preds)):
if targets[j][i] is not None: # Skip those without targets
valid_preds[i].append(preds[j][i].detach())
valid_targets[i].append(targets[j][i])
result = metric_func(valid_targets[i], valid_preds[i])
print('MSE:', result)
fit = plt.figure(figsize=(4,4))
plt.scatter(valid_targets[i], valid_preds[i])
plt.xlabel('Target ClogP')
plt.ylabel('Predicted ClogP')
plt.show()

Almost no correlation as of first epoch. Let's train a few more epochs and see if the prediciton improves.
scheduler = NoamLR(
optimizer=optimizer,
warmup_epochs=[args.warmup_epochs],
total_epochs=[args.epochs] * args.num_lrs,
steps_per_epoch=len(train_data) // args.batch_size,
init_lr=[args.init_lr],
max_lr=[args.max_lr],
final_lr=[args.final_lr]
)
loss_func = nn.MSELoss(reduction='none')
optimizer = Adam(params)
metric_func = mean_squared_error
for epoch in tqdm(range(args.epochs)):
# train
model.train()
loss_sum = iter_count = 0
n_iter = 0
for batch in tqdm(train_data_loader, total=len(train_data_loader), leave=False):
mol_batch, target_batch = batch.batch_graph(), batch.targets()
mask = torch.Tensor([[x is not None for x in tb] for tb in target_batch])
targets = torch.Tensor([[0 if x is None else x for x in tb] for tb in target_batch])
# Run model
model.zero_grad()
preds = model(mol_batch)
# Move tensors to correct device
mask = mask.to(preds.device)
targets = targets.to(preds.device)
class_weights = torch.ones(targets.shape, device=preds.device)
loss = loss_func(preds, targets) * class_weights * mask
loss = loss.sum() / mask.sum()
loss_sum += loss.item()
iter_count += 1
loss.backward()
optimizer.step()
if isinstance(scheduler, NoamLR):
scheduler.step()
n_iter += len(batch)
# eval
model.eval()
preds = []
for batch in tqdm(val_data_loader, disable=False, leave=False):
# Prepare batch
batch: MoleculeDataset
mol_batch = batch.batch_graph()
# Make predictions
with torch.no_grad():
batch_preds = model(mol_batch)
batch_preds = batch_preds.data.cpu().numpy()
# Collect vectors
batch_preds = batch_preds.tolist()
preds.extend(batch_preds)
# valid_preds and valid_targets have shape (num_tasks, data_size)
num_tasks = 1
targets = val_data_loader.targets
valid_preds = [[] for _ in range(num_tasks)]
valid_targets = [[] for _ in range(num_tasks)]
for i in range(num_tasks):
for j in range(len(preds)):
if targets[j][i] is not None: # Skip those without targets
valid_preds[i].append(preds[j][i])
valid_targets[i].append(targets[j][i])
result = metric_func(valid_targets[i], valid_preds[i])
print(epoch, result)
Over the course of training 30 epochs, the MSE quickly improved and reached to less than 0.1 at the end! Let's take a look at the target vs predicted values. So, we got to this level of prediction without ever specifically wrote code for computing ClogP.
fit = plt.figure(figsize=(4,4))
plt.scatter(valid_targets[i], valid_preds[i])
plt.xlabel('Target ClogP')
plt.ylabel('Predicted ClogP')
plt.show()

Conclusion
In this post, I have trained a graph neural network that can predict ClogP property. Within 30 epochs, it was able to predict the property pretty accurately in less than 0.1 MSE. Given only very simple features were used in atom and bond features, it was able to "learn" to predict the property fairly quickly.
Now that we have a trained model, a few things I'd like to try:
- compare this model with other traditional model and compare performance
- try different parameters, such as
depth - try alternative featurization, i.e., add if bond is rotatable in the bond_features and so on.
- add long-range connection;current network is limited to chemical bonds, but longer range interaction may also be important.